PROTOONCOGENES,
ONCOGENES Y ONCOPROTEÍNAS
Cumplen varias
funciones, pero en su conjunto participan en las vías de señalización que
impulsa la proliferación. Ahora bien, los protooncogenes favorecedores del
crecimiento codifican en un estado de normalidad los factores de crecimiento,
los receptores de los factores de crecimiento, transductores de señal, factores
de transcripción o componentes del ciclo celular.
Los oncogenes
correspondientes codifican en general secuencias genómicas que dan como resultado
oncoproteínas que cumplen funciones similares a las de los protooncogenes en
condiciones normales, con la diferencia de que les confieren a dichas proteínas
una autosuficiencia para crecer.
La mayoría de
las neoplasias muestran defectos moleculares de uno o más componentes de estas
vías, a continuación una breve descripción de estos defectos:
Factores de
crecimiento:
Los factores
de crecimiento son sintetizados por un único tipo celular que lo secreta en un
mecanismo paracrino, pero en las células cancerosas adquieren una capacidad
para sintetizar estos factores de crecimiento y mediante una acción autocrina.
Muchos de estos tumores expresan el factor de crecimiento derivado de plaquetas
(PDGF) y la tirosina cinasa de los receptores de PDGF, en otras ocasiones y
dependiendo de la localización del tumor puede que sea el factor de crecimiento
transformante alfa (TGF-alfa) y su receptor asociado, el factor de crecimiento
epidérmico, entre otros. Debemos de tomar en cuenta que el gen propio que sintetiza
el factor de crecimiento no está alterado o mutado, lo que puede que suceda es
que ciertas oncoproteínas determinen la sobreexpresión o la hipersecreción de
estos factores de crecimiento.
Receptores de
los factores de crecimiento:
Varios
oncogenos codifican receptores de factores de crecimiento, en los que destacan
los receptores de la tirosina cinasa. En las células tumorales, los receptores
de la tirosina cinasa se pueden activar de forma constitucional por diversos
mecanismos como mutaciones puntuales, reordenamientos génicos y amplificaciones
génicas.
Algunas
mutaciones de oncogenas de los receptores de factores de crecimiento de
importancia clínica son las siguientes:
ERBB1
Codifica el
receptor del factor de crecimiento epidérmico (EGFR), puntualmente sufre de
mutaciones en algunos adenocarcinomas de pulmón, lo que activa de manera
constitucional la tirosina cinasa EGFR.
ERBB2
Codifica a
otro miembro de la familia de receptores de tirosina cinasa, HER2. Este gen en
determinados tipos de carcinomas, como el de mama, en lugar de tener mutaciones
puntuales sufre de amplificaciones con la consiguiente sobreexposición del
receptor de HER2 y actividad constitucional de la tirosina cinasa.
Reordenamientos
génicos
Estos activan
otros receptores de la tirosina cinasa, como la tirosina cinasa ALK. En este
tipo vemos claramente la deleción del cromosoma 5 que fusiona parte del gen ALK
con parte de otro gen llamado EML4, este es el ejemplo por excelencia en
ciertos adenocarcinomas pulmonares. Este proceso conlleva a la reorganización
de una proteína quimérica EML4-ALK, que posee actividad constitucional de
tirosina cinasa.
Componentes
distales en la vía señalizadora de los receptores de tirosina cinasa: en
condiciones normales el receptor de tirosina cinasa en su porción
citoplasmática estimula la RAS y dos ramas señalizadoras posteriores, la
cascada MAPK y la vía PI3K/AK. Estos componentes sufren de una mutación en
algunos tipos de cáncer, por ejemplo, las mutaciones de RAS, hacen que este se
active y reemplace por completo la función de la actividad de la tirosina
cinasa.
Mutaciones de
RAS
En los humanos
hay tres tipos conocidos HRAS, KRAS, y NRAS; en condiciones normales son miembros
de la familia de pequeñas proteínas G asociadas a la membrana que se unen a
nucleótidos de guanosina (GTP y GDP). Asimismo, se ha demostrado que la RAS
tras una activación transitoria activa una señal de intercambio de GDP por GTP,
lo que estimula las ramas MAPK y PI3K/AKT de la via de señalización de la
tirosina cinasa. En las células cancerosas cuya mutacion es puntual sobre RAS,
reducen notablemente la actividad GTPasa, con esta forma envían señales de
crecimiento constante a las células.
Mutaciones de
oncogenas de BRAF y PI3K
BRAF es una proteína cinasa de serina7treonina
situada en el vértice de una cascada de otras cinasas serina/treonina de la
familia MAPK. Las mutaciones en este activan cada una de estas cinasas y
activan los factores de transcripción. PI3K es un heterodimero compuesto por
una subunidad reguladora y una subunidad catalítica con varias isoformas
tisulares específicas. Las mutaciones de este atacan su subunidad catalítica y
suelen elevar su actividad catalítica.
Alteraciones
de la tirosina cinasa no asociada a receptores
Por lo general estas se
encuentran en el citoplasma o en el nucleo. Estas generan traslocaciones o
reordenamientos cromosómicos que generan genes de difusión que codifican
tirocina cinasa de actividad constitucional.
GENES
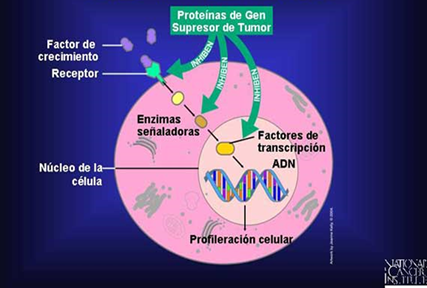
SUPRESORES DE TUMORES
Un gen
supresor tumoral es un gen que reduce la probabilidad de que una célula en un
organismo multicelular se transforme en una célula cancerígena. Los genes
supresores de tumores se encuentran en las células normales y generalmente
inhiben la proliferación celular excesiva. Una mutación o una deleción de un
gen supresor tumoral, aumentará la probabilidad de que se produzca un tumor, al
perder su función. De esa manera, un gen supresor tumoral alterado es similar a
un oncogén.
En las células
normales, las proteínas codificadas por los genes supresores de tumores
detienen la progresión del ciclo celular en respuesta a un daño en el ADN o a
señales de supresión del crecimiento provenientes del medio extracelular.
Cuando los genes supresores de tumores están mutados o son inactivos, las
células no pueden responder normalmente a los puntos de control del ciclo
celular, o son incapaces de realizar muerte celular programada si el daño del
ADN es demasiado importante.
Funciones de
los productos de los genes supresores de tumores:
Las proteínas
codificadas por la mayoría de los genes supresores de tumores inhiben la
proliferación o la supervivencia de la célula. Por lo tanto, la inactivación de
los genes supresores de tumores conduce al desarrollo del tumor eliminando
proteínas de regulación. Varios genes supresores de tumores codifican proteínas
reguladoras de transcripción. Otros de los productos de estos genes regulan la
progresión del ciclo celular siendo capaces de actuar como oncogenes. Los
productos de estos genes actúan a través de mecanismos muy diversos:
Inhibiendo la
progresión de las células a través del ciclo celular.
Haciendo que
las células entren en apoptosis.
Manteniendo la
estabilidad del genoma (replicación, reparación y segregación).
Los genes
supresores de tumores pertenecen a distintos tipos de proteínas, como factores
de crecimiento, de adhesión celular, control del ciclo celular, factores de
transcripción, reparación del ADN
Actualmente se
conocen tres formas de inactivación de estos genes:
Por mutaciones
puntualesLas más
frecuentes son las que conducen a cambios en el marco de lectura del gen,
mutaciones que conducen a codones de parada (codones STOP) y mutaciones de
cambio de aminoácido.
Por deleción
Con
frecuencia, la deleción incluye además a genes vecinos del gen supresor de
tumor.
Por
metilación.
Tipos de genes supresores:
Genes
supresores de tumores “guardianes o gatekeepers”
Se encuentran
en los síndromes de cáncer hereditario autosómico dominantes. Su función es
regular directamente el crecimiento celular. Bloquean el desarrollo de tumores
al regular la transición de las células a través de los puntos de control
existentes en el ciclo celular o mediante la estimulación de la muerte celular
programada, con control de la división y la supervivencia celulares. Las
mutaciones con pérdida de función en los genes guardianes dan lugar a una
mutación celular incontrolada.
Los genes
supresores de tumores guardianes codifican:
- Los reguladores de diversos puntos de
control del ciclo celular.
- Los mediadores de la muerte celular
programada.
Genes
“cuidadores” o “de mantenimiento” (caretakers)
Se encuentran
en los síndromes de cáncer autosómico dominante. Están implicados en la
reparación de las alteraciones del ADN y en el mantenimiento de la integridad
del genoma. La pérdida de función de los genes cuidadores permite la
acumulación de mutaciones en los oncogenes y en los genes guardianes, lo que da
lugar, en conjunto, a la iniciación y la promoción del cáncer.
Los genes
supresores de tumores cuidadores codifican:
- Las proteínas responsables de la
detección y la reparación de las mutaciones.
- Las proteínas implicadas en las
disyunciones cromosómicas normales durante la mitosis.
- Los componentes del dispositivo de
muerte celular programada.